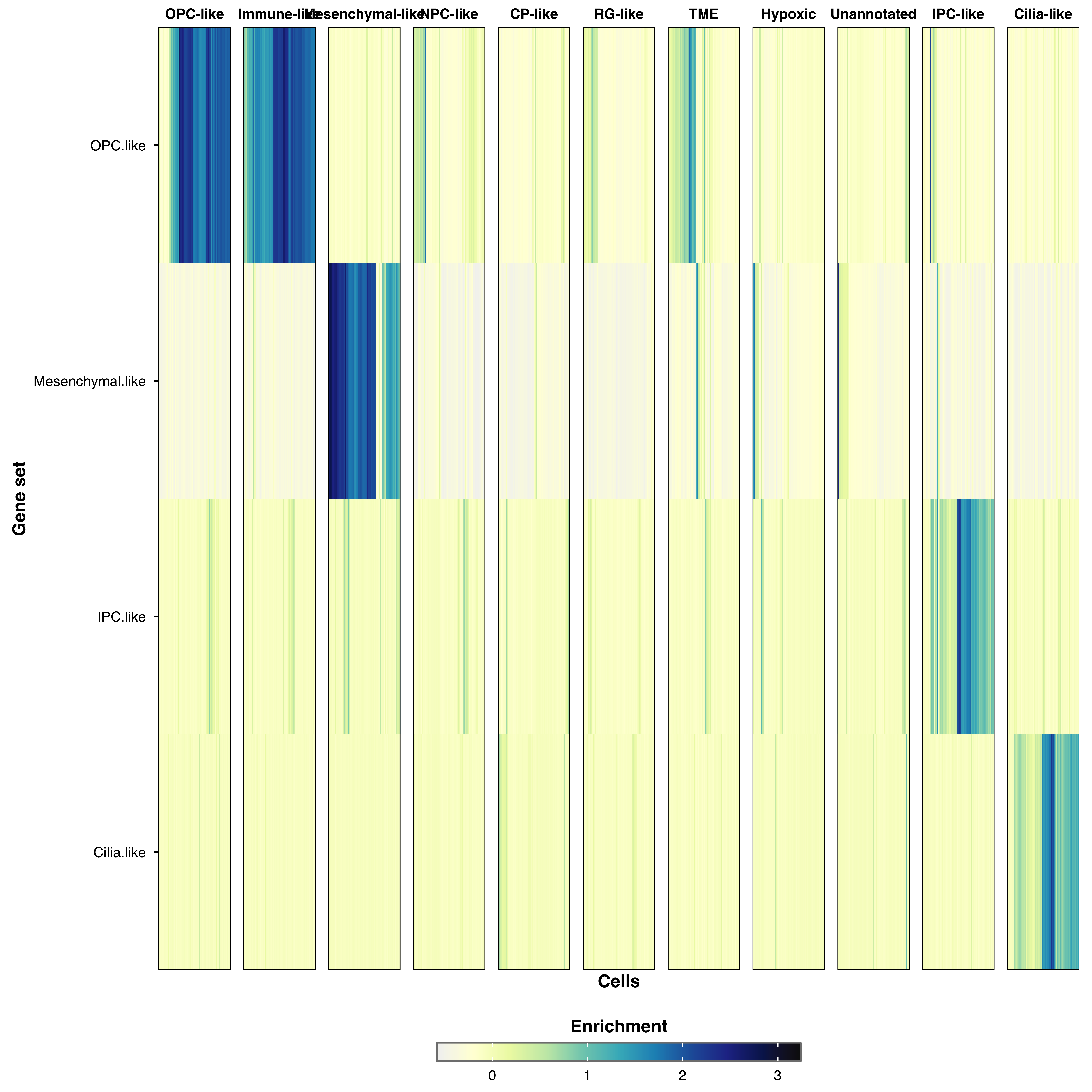
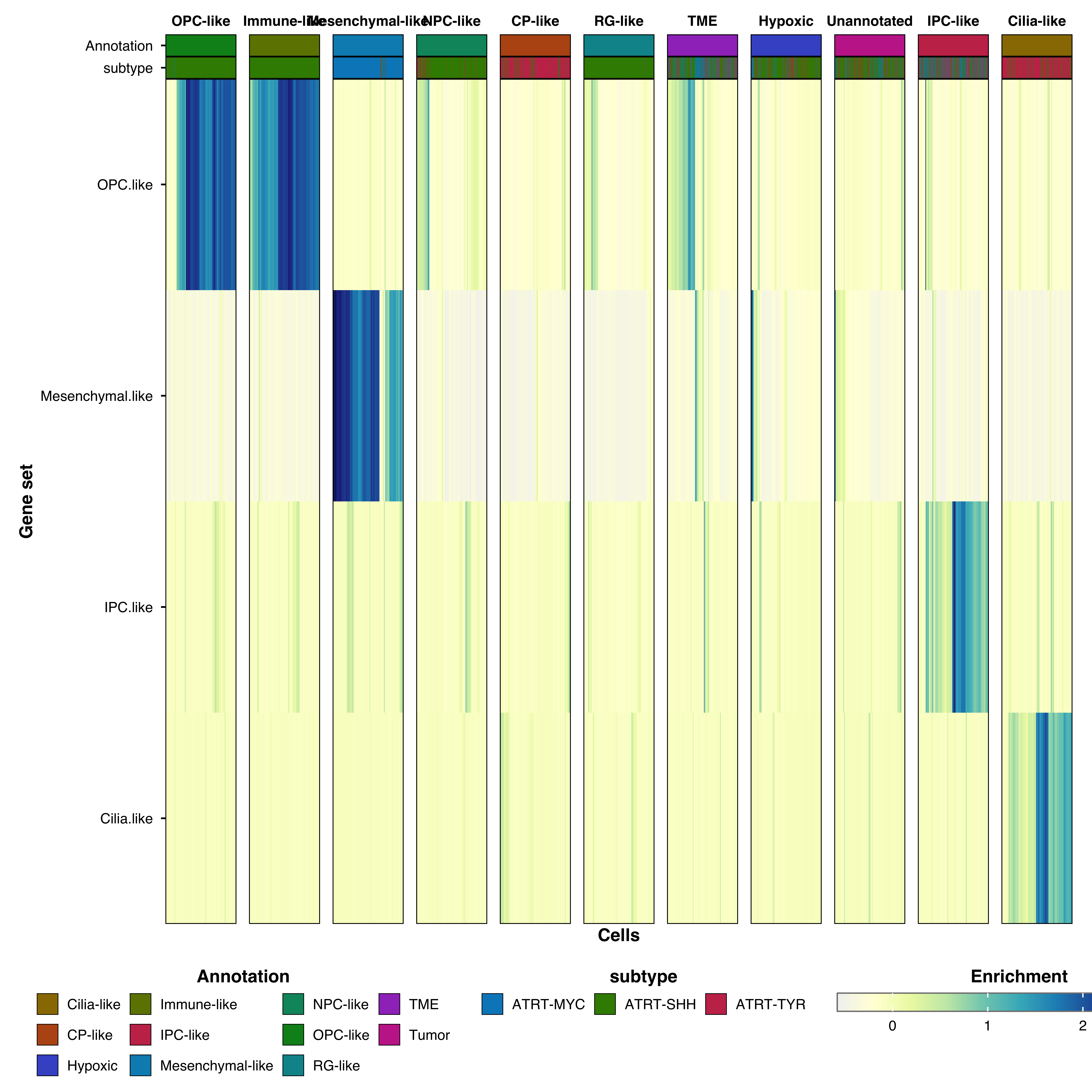
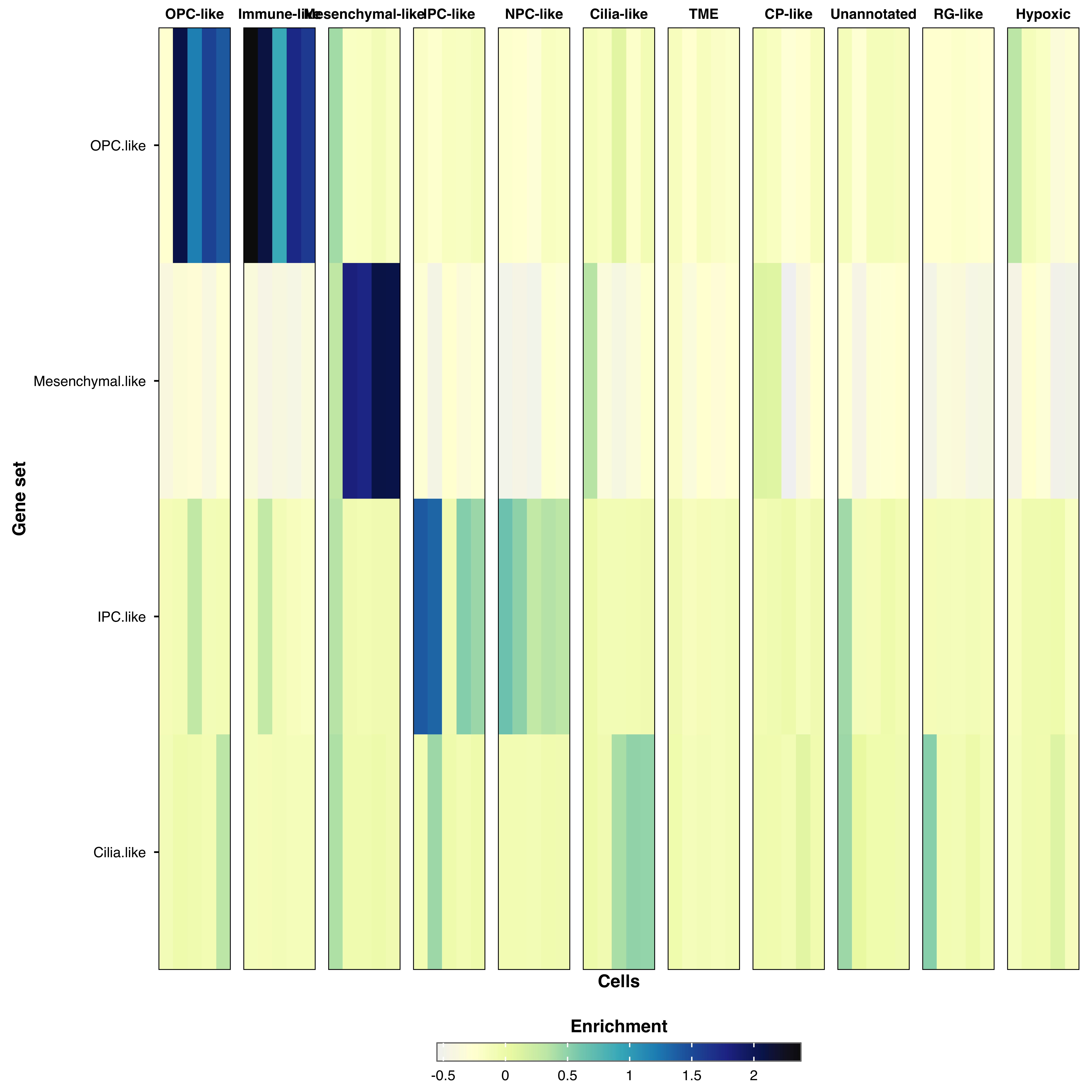
Single-cell enrichment heatmaps display enrichment scores at individual cell resolution . Similar to do_SCExpressionHeatmap but for gene set scores instead of individual genes.
Basic usage
gene_lists <- list ( "IPC.like" = c ( "CDC25C" , "KIF18B" , "KIF14" , "CENPE" ) ,
"Cilia.like" = c ( "DNAAF1" , "ADGB" , "CFAP61" , "CFAP157" ) ,
"OPC.like" = c ( "KCNQ5" , "MEOX2" , "MBP" , "SNTG1" ) ,
"Mesenchymal.like" = c ( "S100A1" , "MGP" , "TNNT1" , "H2AFJ" )
) p <- SCpubr :: do_SCEnrichmentHeatmap ( sample = sample , input_gene_list = gene_lists )
p
Scoring methods
Use flavor = "Seurat" or flavor = "UCell" to toggle different scoring methods.
Return enriched object
result <- SCpubr :: do_SCEnrichmentHeatmap ( sample = sample , input_gene_list = gene_lists ,
return_object = TRUE )
p <- result $ plot sample_enriched <- result $ object
Parameter reference
For parameters shared across many functions (color palettes, typography, legend styling), see Shared features .
Core parameters
input_gene_listNamed list of gene signatures
—
Scoring method
flavor
"Seurat" or "UCell"
"Seurat"
nbinBins for Seurat scoring
24
ctrlControl genes per bin
100
ncoresCPU cores for UCell
1
Layout
proportional.sizeEqual space per group
TRUE
main.heatmap.sizeMain heatmap proportion
0.95
subsampleMax cells per group
NA
clusterCluster gene sets
TRUE
return_objectReturn Seurat object
FALSE