27 Azimuth reference mapping reports
Reference mapping has become a very handy tool in our disposal to assess the identity of our clusters. This normally works by using a dataset as a reference (normally, a single-cell atlas), and projecting our cells onto the atlas. Out of the many tools available to perform such analyses, Azimuth is one worth noting. Developed by the Seurat team, it perfectly integrates and works with Seurat objects.
Running Azimuth requires some preparation, and the results can be a bit complex to interpret at first glance. To cover this need, one can use SCpubr::do_AzimuthAnalysisPlot(). This function requires as input the output Seurat object resulting of running Azimuth in R (not in their interactive website). Here is a piece of code to get a suitable input:
library(Azimuth)
library(Seurat)
# Seurat object
sample <- your_seurat_object
# Consult available references.
SeuratData::AvailableData()
# Install the reference of interest.
ref = "pbmc3k"
SeuratData::InstallData(ref)
# Load the dataset.
library(pbmcref.SeuratData)
# Metadata column from the reference containing the annotation we want to infer from our sample.
annotation.levels <- "predicted.celltype.l1" # This is dependent on the reference.
# Compute Azimuth mapping.
sample <- Azimuth::RunAzimuth(query = sample,
reference = "pbmcref",
annotation.levels = annotation.levels)
# The sample is ready for running SCpubr::do_AzimuthAnalysisPlot()!27.1 Basic analysis
The basic analysis that can be carried out from Azimuth’s output goes as follows:
Azimuth provides an annotation score as well as a mapping score. Both have a meaning and are perfectly explained in their website. From there:
Prediction scores: Cell prediction scores range from 0 to 1 and reflect the confidence associated with each annotation. Cells with high-confidence annotations (for example, prediction scores > 0.75) reflect predictions that are supported by mulitple consistent anchors. Prediction scores can be visualized on the Feature Plots tab, or downloaded on the Download Results tab. The prediction depends on the specific annotation for each cell. Therefore, if you are mapping cells at multiple levels of resolution (for example level 1/2/3 annotations in the Human PBMC reference), each level will be associated with a different prediction score.
Mapping scores: This value from 0 to 1 reflects confidence that this cell is well represented by the reference. The “mapping.score” column is available to plot in the Feature Plots tab, and is provided in the download TSV file. The mapping score is independent of a specific annotation, is calculated using the MappingScore function in Seurat, and reflects how well the unique structure of a cell’s local neighborhood is preserved during reference mapping.
This means, that depending on how strict we want to be in our analyses, we can set a cutoff for either of the scores (or both). This is defined in SCpubr::do_AzimuthAnalysisPlot() by the parameters mapping.cutoff (for the mapping scores) and annotation.cutoff (for the annotation scores). The cells that do not surpass the cutoffs will turn into NA for the plotting, as Azimuth will always give a labelling to each of the cells regardless of the mapping and annotation scores.
Then, in order to run SCpubr::do_AzimuthAnalysisPlot() one needs to provide:
- The Seurat object that has undergone
Azimuth::RunAzimuth(). - The name of the metadata column that stores the annotation (to
annotation.labels). - The name of the metadata column that stores the annotation scores (to
annotation.scoring). - The name of the metadata column that stores the mapping scores (to
mapping.scoring). - The cutoff for the annotation scores (0.75 by default).
- The cutoff for the mapping scores (0 by default).
As follows:
# Generate an Azimuth report.
out <- SCpubr::do_AzimuthAnalysisPlot(sample = sample,
annotation.labels = "predicted.celltype.l1",
annotation.scoring = "predicted.celltype.l1.score",
font.size = 18)By default, it will generate a list of objects, that contain:
- A vector of the resulting annotation (to use or store).
- A FeaturePlot of the mapping scores.
- A FeaturePlot of the annotation scores.
- A DimPlot of the current annotation (or whatever metadata annotaiton provided to
group.by). - A DimPlot of the inferred annotation.
- A DimPlot of our queried cells in the reference UMAP.
- A stacked BarPlot showing the proportion of the different datasets of origin (stored in
orig.ident) across the current annotation. This is specially useful in merged tumor datasets in which the tumor microenvironment clustes will merge together in single clusters while the tumor cells from each individual datasets will form single clusters. Otherwise, this plot will be very little informative. - A stacked BarPlot showing the proportion of the inferred annotation in each of our original clusters.
- A report containing all of the above plots in landscape and portrait formats.
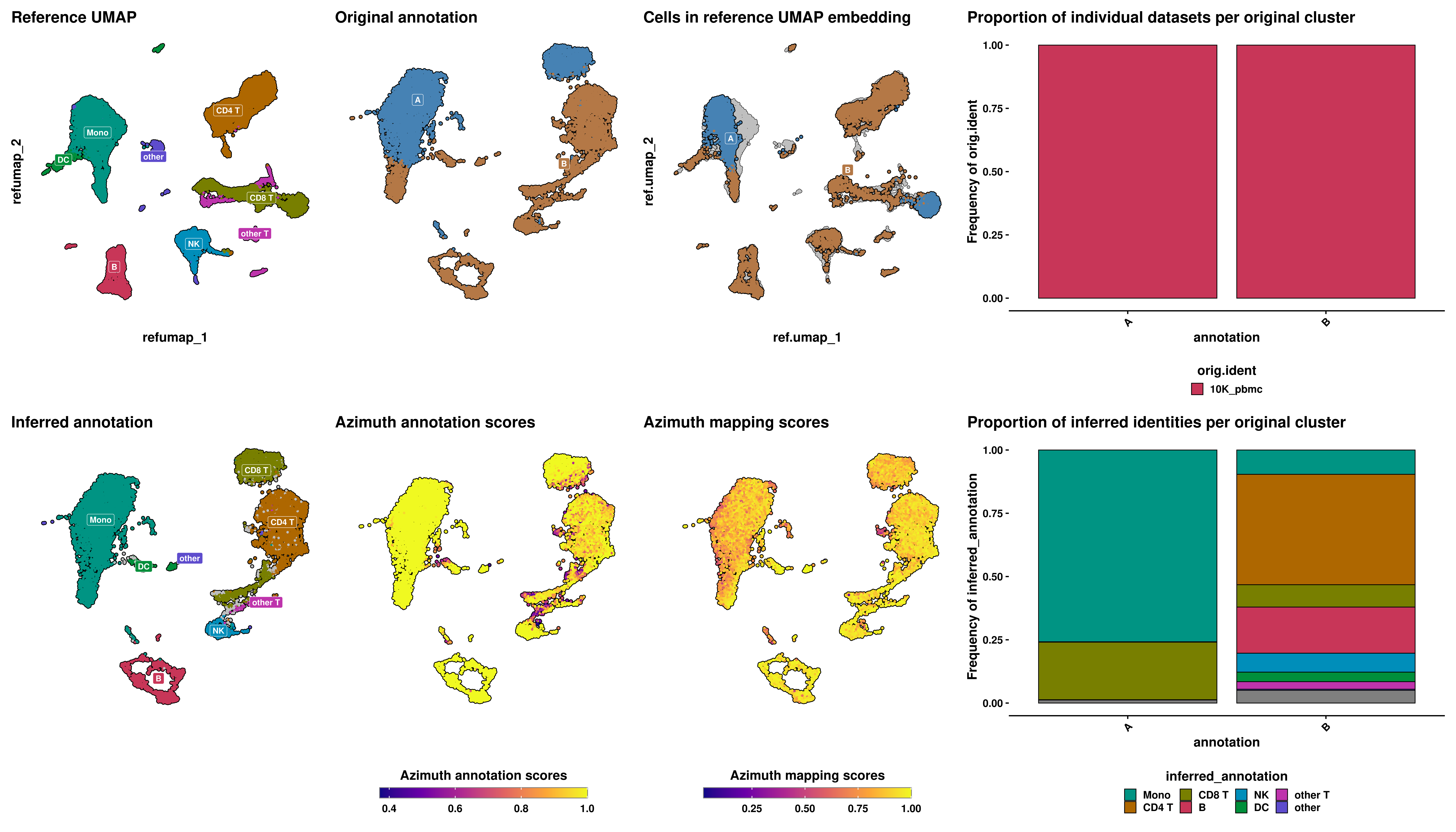
Let’s have a look at the report:
# Azimuth report - portrait.
out$report_portrait
# Azimuth report - landscape.
out$report_landscape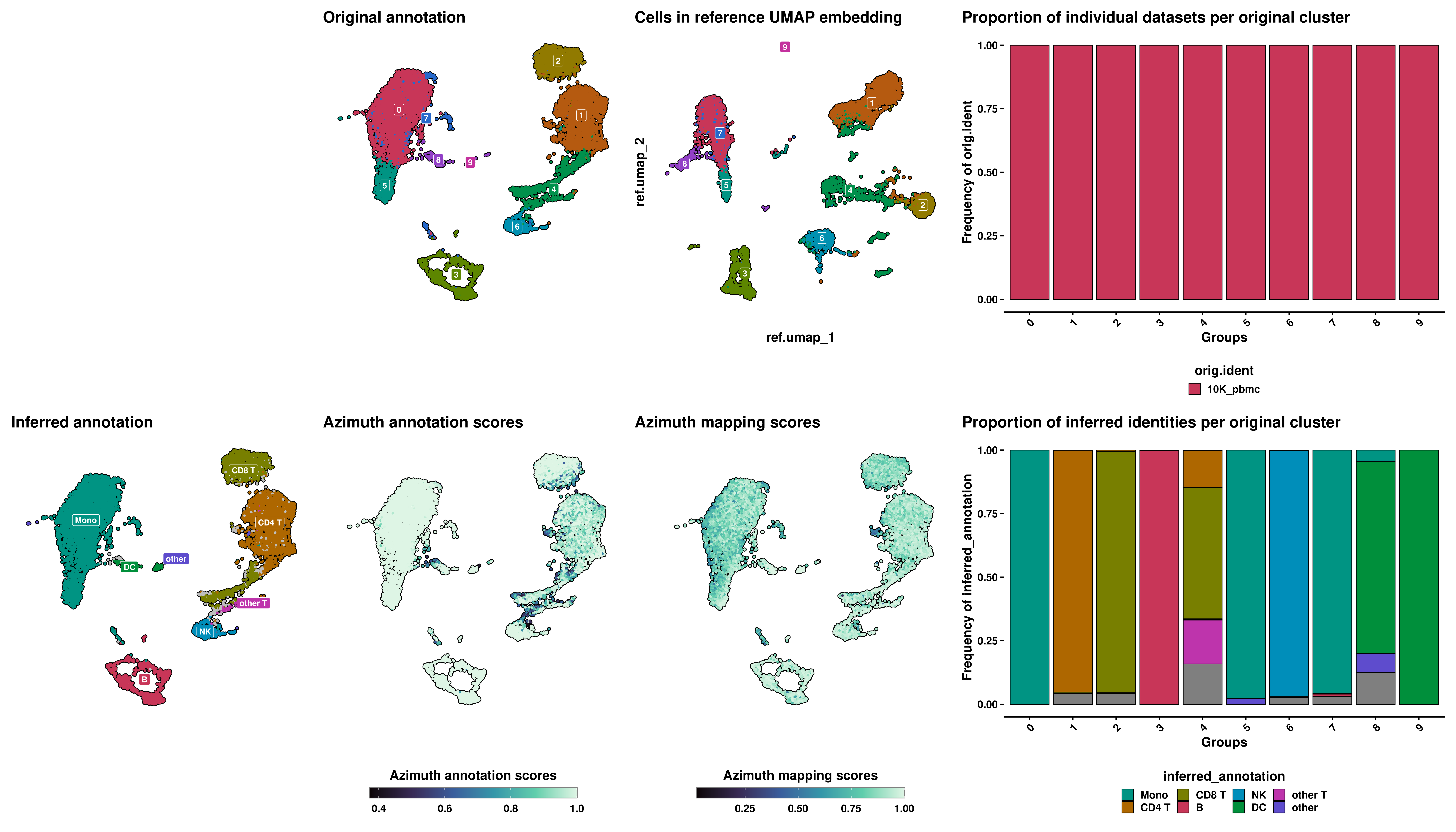
27.2 Add the reference dataset to the report
As can be observed, there is an empty space in the report. This is meant to be filled by a UMAP of the original reference, if we happen to have the original object at hand. In the cases of the datasets installed using SeuratData, one can retrieve them as:
# Retrieve reference.
reference <- readRDS(system.file("azimuth/ref.Rds", package = "pbmcref.SeuratData"))
# Set the identities of the object to match our inferred identities.
Seurat::Idents(reference) <- reference$celltype.l1 The path to this object varies from package to package and also depends on the user’s installation. It is highly recommendable to navigate through your installation folder where all R packages are installed and search for the object manually.
Then, we can provide the reference object using ref.obj parameter and it is also highly recommended to provided the UMAP dimensional reduction name, which in this case is refUMAP, but also can vary from dataset to dataset. The names can be checked using Seurat::Reductions(sample) and the name has to be fed to ref.reduction parameter, as such:
# Generate an Azimuth report with the reference object.
out <- SCpubr::do_AzimuthAnalysisPlot(sample = sample,
annotation.labels = "predicted.celltype.l1",
annotation.scoring = "predicted.celltype.l1.score",
ref.obj = reference,
ref.reduction = "refUMAP",
font.size = 18)# Azimuth report - portrait with reference.
out$report_portrait
# Azimuth report - landscape with reference.
out$report_landscape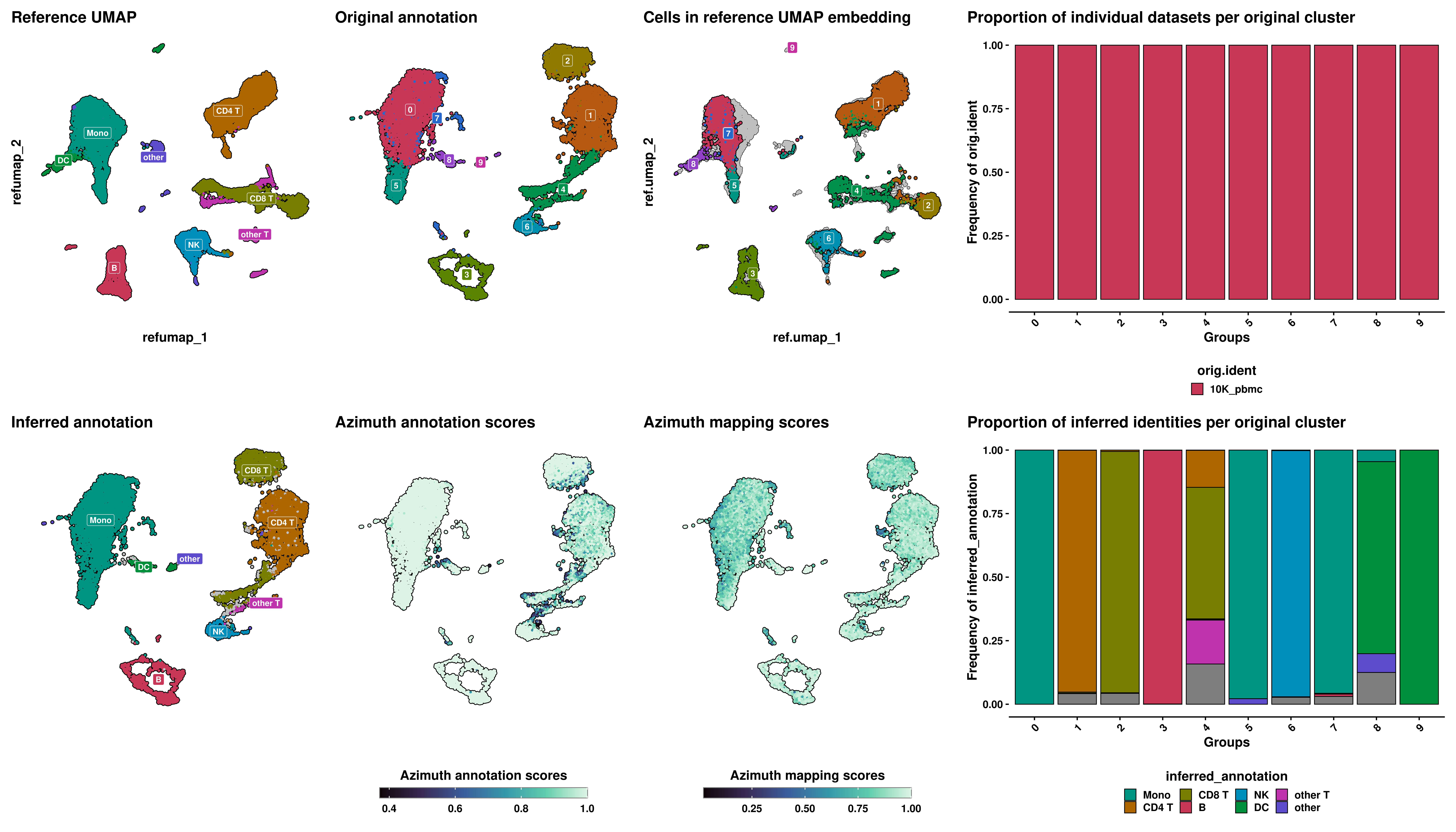
As can be observed, a new DimPlot with the original UMAP is now displayed at the top left of the report. What is more interesting, since now we have the original UMAP coordinates, we can also project the UMAP silhouette onto the UMAP where our cells in the reference UMAP were shown, so we have a better understanding of where exactly in the UMAP they are actually located.
27.3 Use custom grouping
As usual, we can make use of group.by parameter to dictate which identities are being used for plotting:
# Generate an Azimuth report with a custom grouping.
sample$annotation <- ifelse(sample$seurat_clusters %in% c("0", "2", "7"), "A", "B")
out <- SCpubr::do_AzimuthAnalysisPlot(sample = sample,
group.by = "annotation",
annotation.labels = "predicted.celltype.l1",
annotation.scoring = "predicted.celltype.l1.score",
ref.obj = reference,
ref.reduction = "refUMAP",
font.size = 18)# Azimuth report - portrait with reference and custom grouping.
out$report_portrait
# Azimuth report - landscape with reference and custom grouping.
out$report_landscape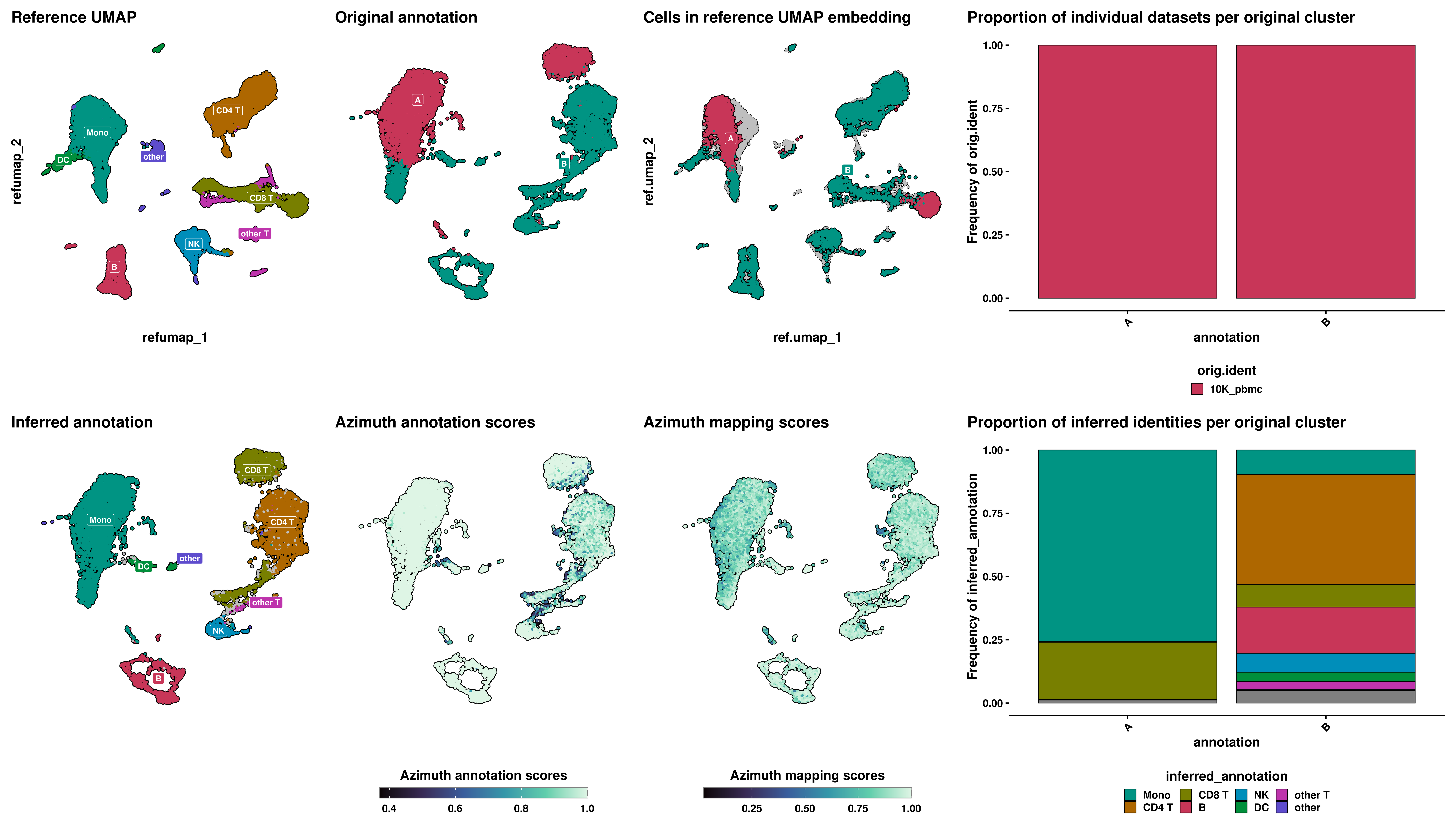
27.4 Use custom colors
Finally, as usual, we can provide our custom color scheme to the DimPlots using colors.use and to the FeaturePlots using viridis_color_map and viridis_direction:
# Generate an Azimuth report with a custom grouping.
colors.use <- SCpubr::do_ColorPalette("steelblue", opposite = TRUE)
names(colors.use) = c("A", "B")
out <- SCpubr::do_AzimuthAnalysisPlot(sample = sample,
group.by = "annotation",
annotation.labels = "predicted.celltype.l1",
annotation.scoring = "predicted.celltype.l1.score",
ref.obj = reference,
ref.reduction = "refUMAP",
colors.use = colors.use,
viridis_color_map = "C",
viridis_direction = 1,
font.size = 18)# Azimuth report - portrait with reference and custom colors.
out$report_portrait
# Azimuth report - landscape with reference and custom colors.
out$report_landscape