# Seurat's dot plot.
p <- Seurat::DotPlot(sample,
features = "CD14")
p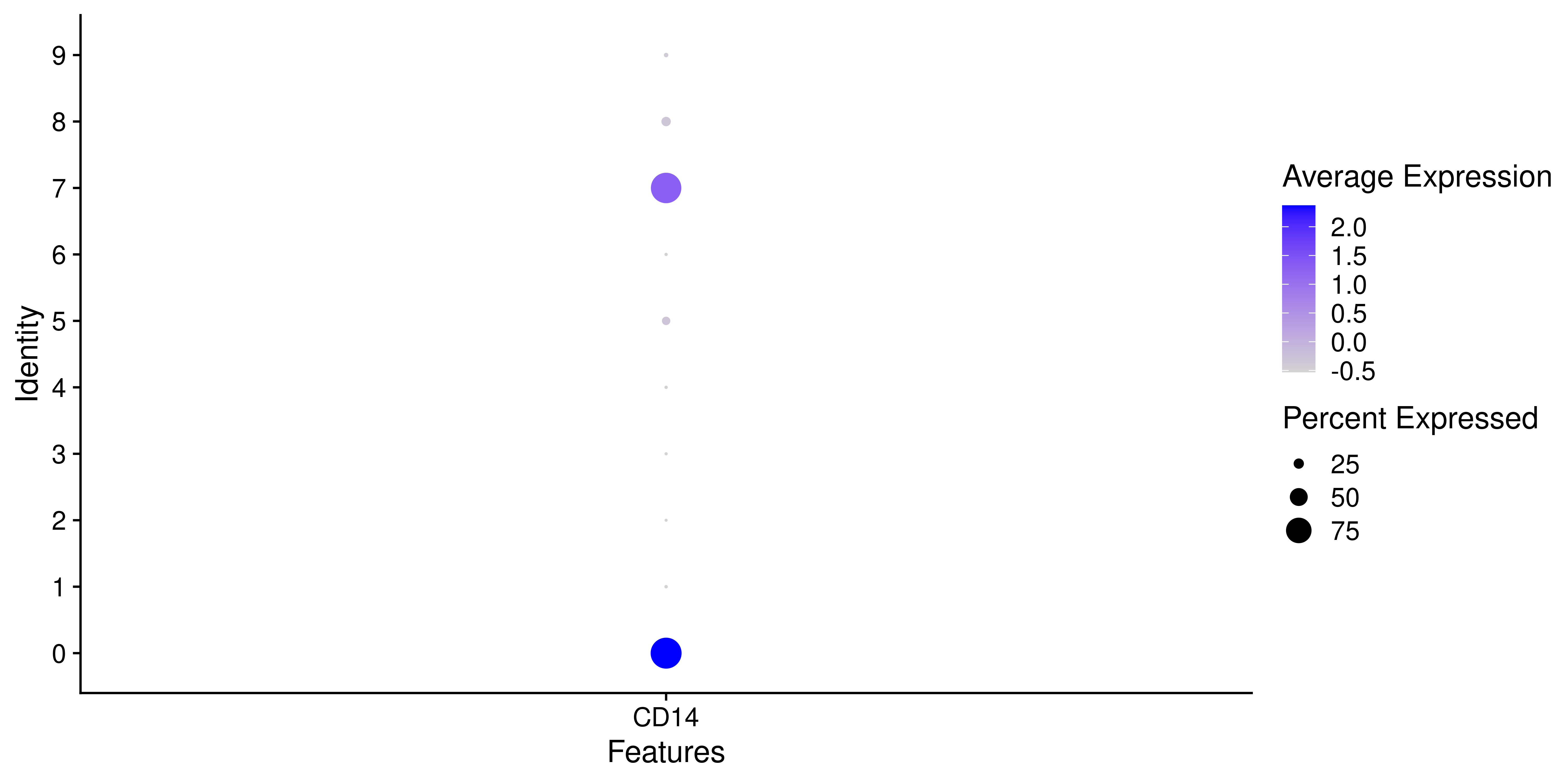
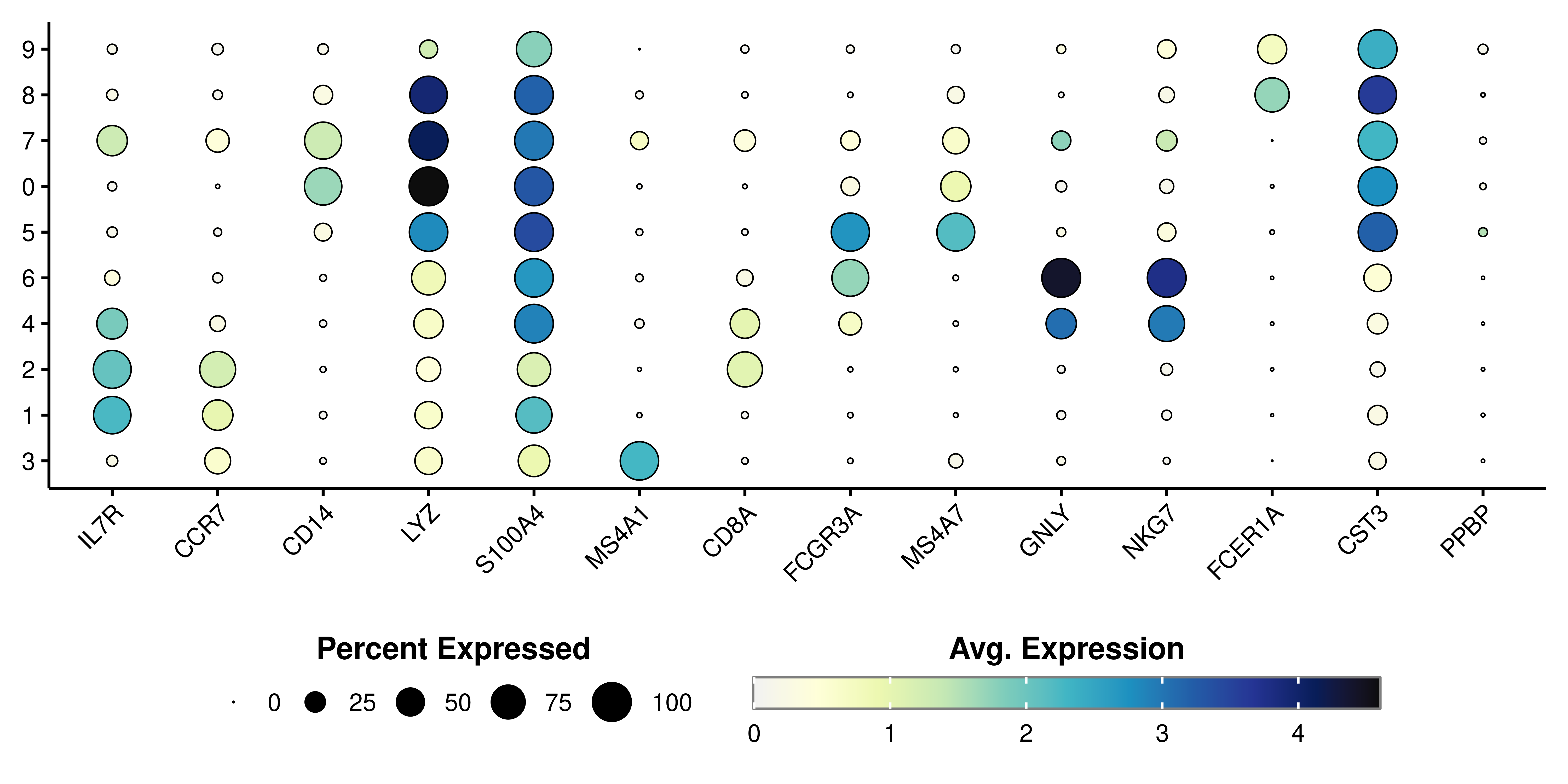
Dot plots are a very nice data representation technique. It involves the use of two scales. One scale is color-based while the second one is size-based. In the context of Seurat::DotPlot(), these scales are pre-defined to contain the average expression values on the color scale and the percentage of cells within the group expressing the feature on the size, increasing it as this percentage grows bigger. It is heavily implied that the features need to be genes, otherwise the color scale will be lost. This is the output from Seurat:
# Seurat's dot plot.
p <- Seurat::DotPlot(sample,
features = "CD14")
p
As can be seen, if we try to plot other type of variables, the resulting plot will not contain a color scale. SCpubr::do_DimPlot() focuses on enhancing the overall appearance of the plots. Implementation using other types of features is not currently supported. This is how it looks using SCpubr::do_DimPlot():
# SCpubr's dot plot.
p <- SCpubr::do_DotPlot(sample = sample,
features = "CD14")
p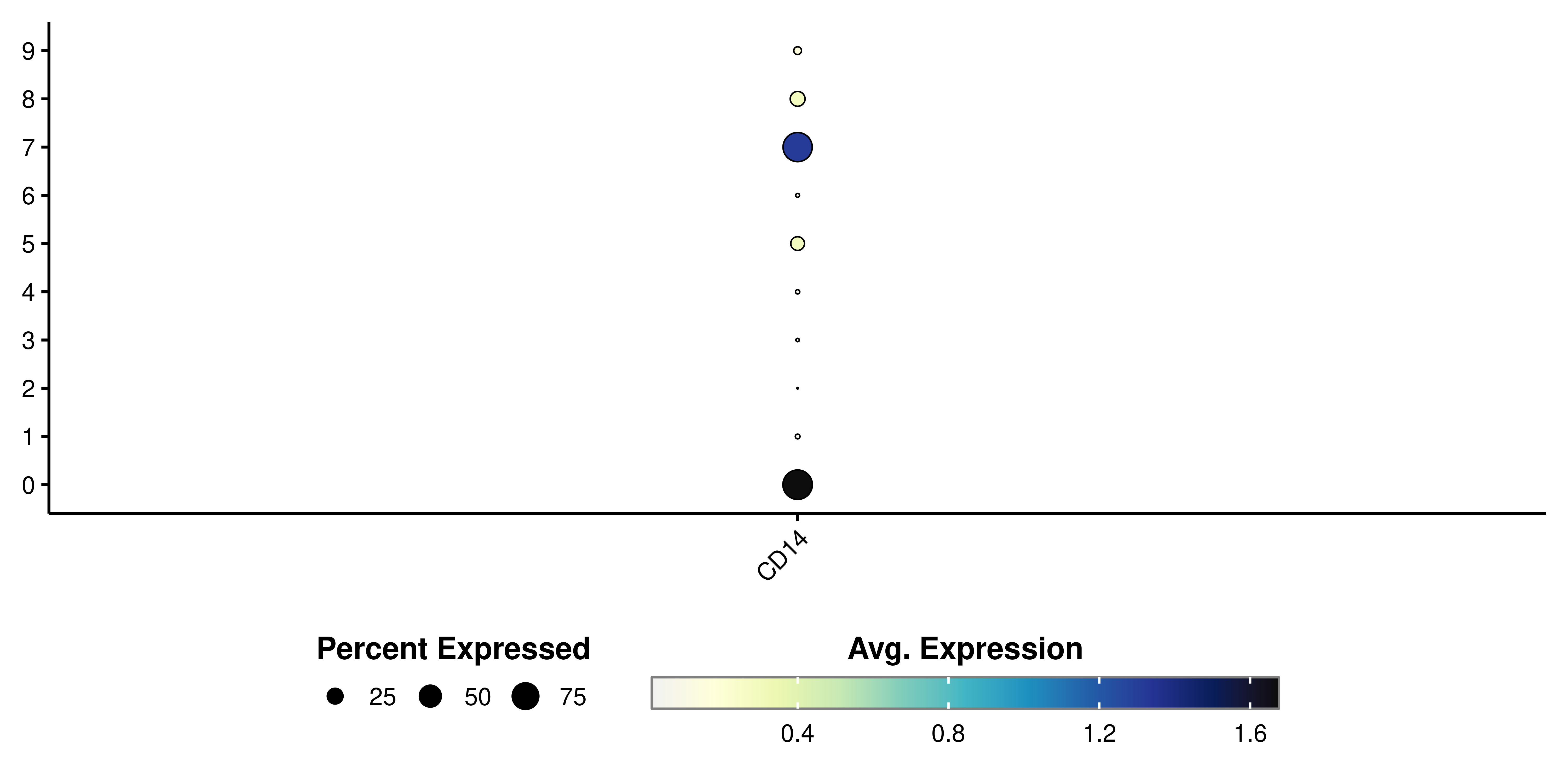
The true power of this data visualization relies on querying multiple genes at once. Let’s query the ones described in this Seurat vignette:
genes <- c("IL7R", "CCR7", "CD14", "LYZ",
"S100A4", "MS4A1", "CD8A", "FCGR3A",
"MS4A7", "GNLY", "NKG7", "FCER1A",
"CST3", "PPBP")
p <- SCpubr::do_DotPlot(sample = sample,
features = genes)
p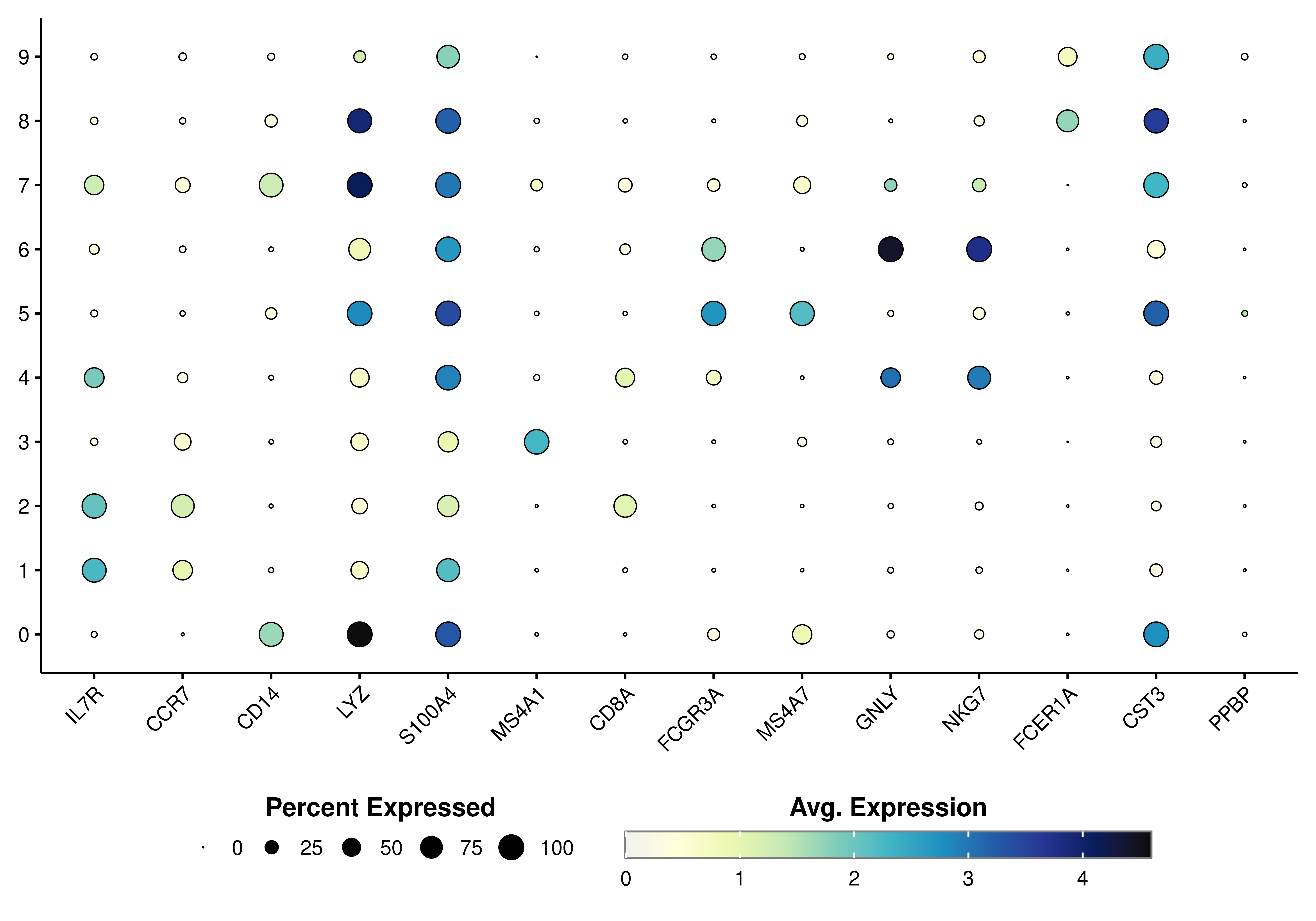
Here, we can clearly distinguish which clusters are highly enriched in which given genes. This proves to be a very interesting way to query for cluster identities. It is highly dependent, though, on the accuracy of the selected genes for the queried dataset. If we actually provide the list of genes as a named list, we can facet the plot in the different list of genes provided:
genes <- list("Naive CD4+ T" = c("IL7R", "CCR7"),
"CD14+ Mono" = c("CD14", "LYZ"),
"Memory CD4+" = c("S100A4"),
"B" = c("MS4A1"),
"CD8+ T" = c("CD8A"),
"FCGR3A+ Mono" = c("FCGR3A", "MS4A7"),
"NK" = c("GNLY", "NKG7"),
"DC" = c("FCER1A", "CST3"),
"Platelet" = c("PPBP"))
p <- SCpubr::do_DotPlot(sample = sample,
features = genes)
p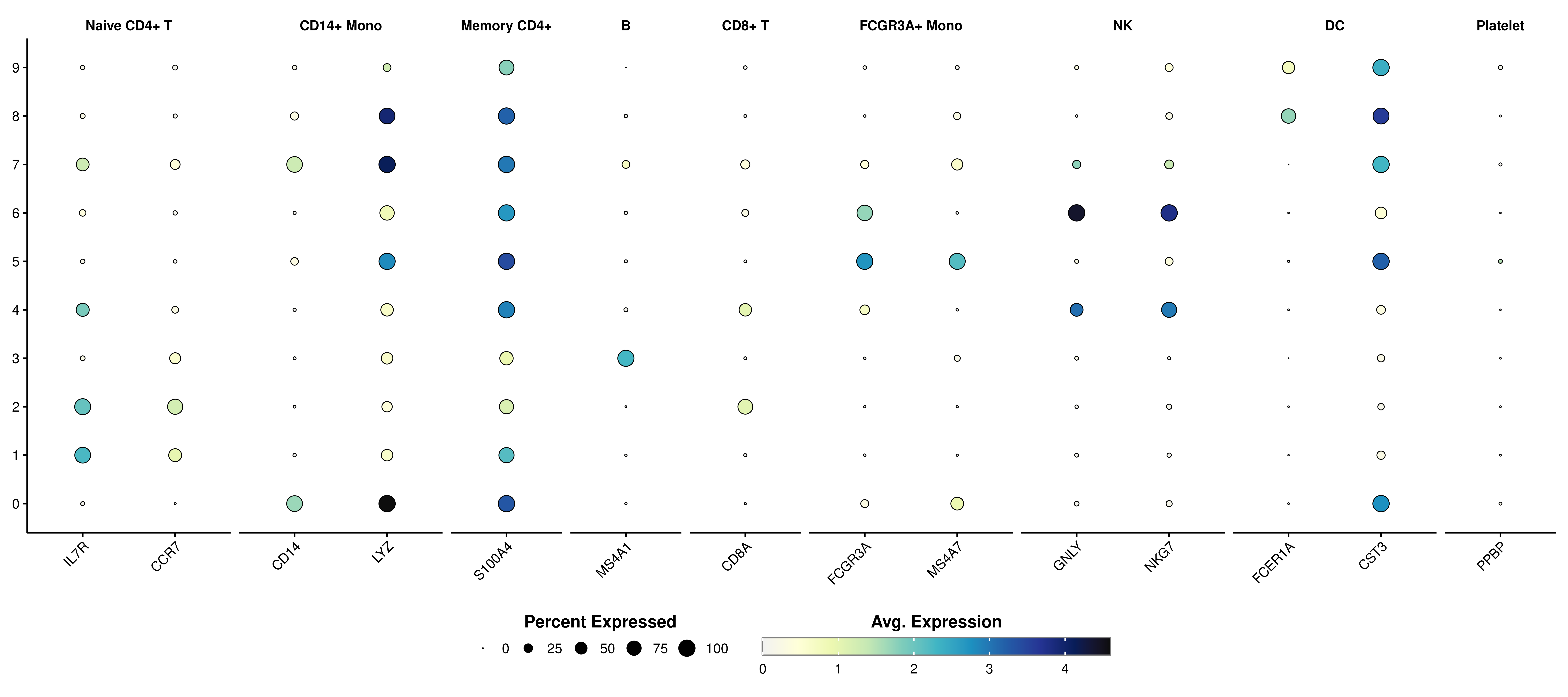
One really cool feature from Seurat::DotPlot() is that it lets you cluster the identities in the Y axis depending on how similar they are between them across the values on the X axis. This is achieved by using cluster = TRUE, parameter also implemented in SCpubr::do_DotPlot():
p <- SCpubr::do_DotPlot(sample = sample,
features = genes,
cluster = TRUE)
p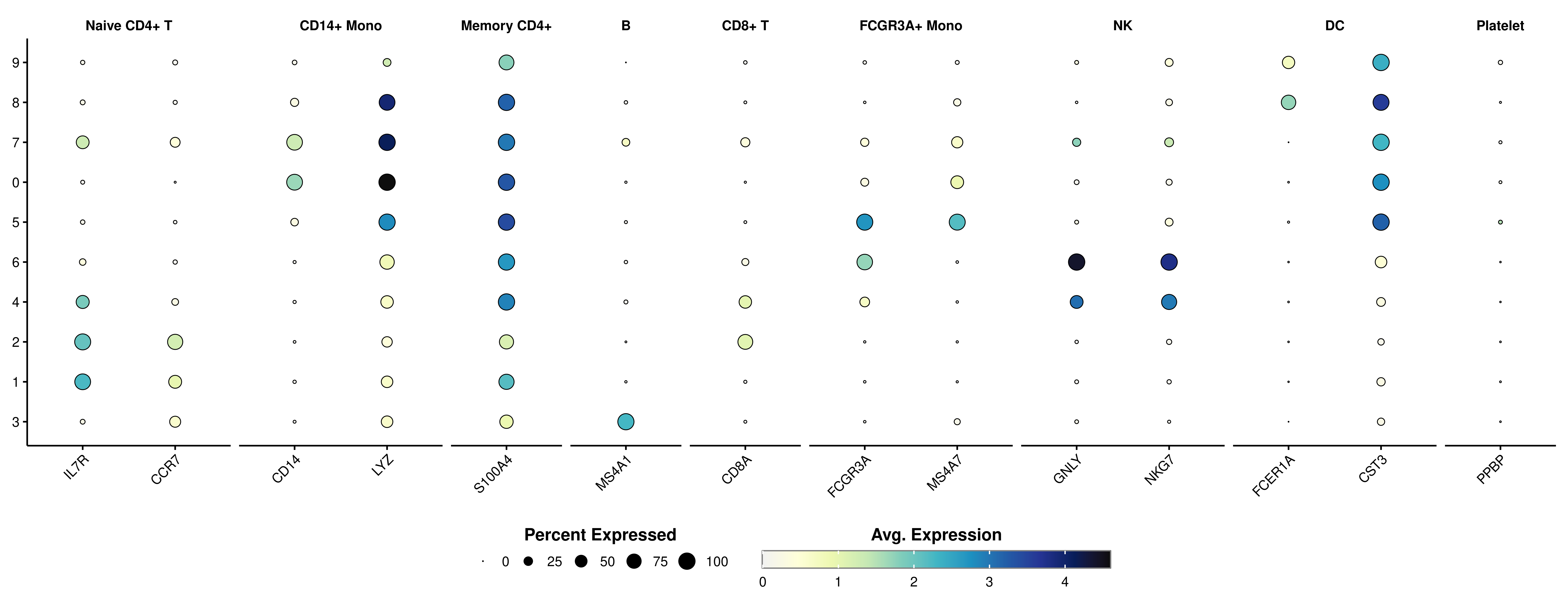
You can increse the dot size with dot.scale parameter.
genes <- c("IL7R", "CCR7", "CD14", "LYZ",
"S100A4", "MS4A1", "CD8A", "FCGR3A",
"MS4A7", "GNLY", "NKG7", "FCER1A",
"CST3", "PPBP")
p <- SCpubr::do_DotPlot(sample = sample,
features = genes,
cluster = TRUE,
dot.scale = 8)
p